Galerija

Evropski trg medicinske tehnologije je vreden 140 milijard evrov in predstavlja 27,6 odstotka svetovnega trga. Pomembna prelomnica v delovanju celotnega sektorja EU na področju medicinskih pripomočkov je bila nova uredba o medicinskih pripomočkih 2017/745 (MDR), ki je začela veljati 26. maja lani. In kaj o učinkih novih pravil lahko rečemo po dobrem letu dni?
Nova pravila morajo od omenjenega datuma izpolnjevati vsi medicinski pripomočki, ne glede na razred tveganja, najdlje do 26. maja 2024 pa proizvajalci lahko dajejo na trg že certificirane izdelke po prejšnji direktivi MDD, če se zasnova in predvideni namen medicinskega pripomočka nista bistveno spremenila. Certifikati po MDD po 26. maju 2024 ne bodo več veljavni, kar pomeni, da bodo morali vsi, ki že imajo proizvode na trgu, ponovno izvesti postopek certifikacije, pojasnjuje Ana Pribaković Borštnik, pomočnica direktorja področja Ocenjevanje sistemov vodenja za področje medicinskih pripomočkov v SIQ. Pripomočke, ki bodo pred 26. majem 2024 prišli na trg distributerjev in še ne bodo šli do končnega uporabnika, pa distributerji lahko prodajajo še leto dni dlje, do 26. maja 2025.
Slovenski institut za kakovost in meroslovje (SIQ Ljubljana) je marca letos postal priglašeni organ po uredbi MDR ter tako edina institucija v širši regiji, ki slovenskim proizvajalcem lahko omogoča tako širok nabor storitev s področja preverjanja medicinskih pripomočkov na enem mestu. Priglašenih organov po MDR je precej manj kot prej, ko jih je bilo 50. Sedaj jih je le 31. SIQ kot priglašeni organ po novi uredbi omogoča, da medicinske pripomočke, ki so ključni pri reševanju življenj, varno in skladno z zakonodajo lahko uporabljamo na enotnem evropskem trgu.
Pri medicinskih pripomočkih bo treba ponovno izvesti postopke certifikacije, kar pomeni ponoven pregled tehnične dokumentacije in ponovne presoje na lokaciji skladno z zahtevami nove uredbe. »Pri novi uredbi je velik poudarek na dokazovanju učinkov delovanja medicinskih pripomočkov. Poleg tega je nekaj novih zahtev glede varnosti in učinkovitosti, precej poudarka pa je tudi na programski opremi in njeni varnosti, vključno s kibernetsko varnostjo, saj je v medicinskih pripomočkih vse več programske opreme in prenosa podatkov, tudi prek mobilnih telefonov, kar je še dodatno tveganje. Dokler si na mrežah v svojih zaprtih sistemih, je drugače, kot če si nenehno na udaru prek mobilnih telefonov. Pravi hit je namreč postala uporaba medicinske programske opreme na mobilnih telefonih,« je še pojasnila Ana Pribaković Borštnik.

Na SIQ dobijo zelo veliko vprašanj od proizvajalcev, ki imajo svoje medicinske pripomočke že na trgu, kakšen postopek jih čaka zaradi nove uredbe, koliko časa bo potekal in kakšen strošek bo zanje s tem nastal. »Je pa tudi veliko novih proizvajalcev, ki želijo prenesti certifikat ali pa se na novo certificirati pri nas, bodisi z novimi bodisi s sedanjimi proizvodi. Verjetno je zato, ker so priglašeni organi tako zasedeni, tudi njihovi sedanji.« Gotovo pa je k temu pripomoglo tudi dejstvo, da je treba veliko več stvari preveriti, saj se zdaj zahteva enako natančen pregled tehnične dokumentacije ne glede na to, ali gre za proizvod višjega ali nižjega tveganja. Sicer pa je za vsak medicinski pripomoček treba v živo na lokaciji pogledati, kako se proizvaja, kako je v resnici videti.
To seveda pomeni več ur dela in seveda višji strošek za proizvajalca. In kot še pravi sogovornica, se zaradi tega lahko zgodi, da bodo manjši proizvajalci ali pa tisti, ki prodajo manj medicinskih pripomočkov, ali prenehali proizvajati ali pa bodo prodali svoje pripomočke. Gre za tiste, ki so bili že po MDD na meji finančne vzdržnosti svojega proizvoda, po novi uredbi pa se lahko zgodi, da tega ne bodo zdržali.
Vendar po drugi strani nova pravila prinašajo tudi večjo varnost za potrošnika. »Spremembe, ki jih je uvedla uredba, so bile potrebne, to je poenotenje meril in strožja merila za posamezne zahteve. Pomembna pa je tudi dostopnost do pripomočkov in inovativnih rešitev, pri čemer je vedno treba najti kompromis. V zakonodaji so opredeljene zahteve, praksa pa potem pokaže, kako se zahteve zares uveljavijo – kje je treba vztrajati, ker je zaradi varnosti in učinkovitosti to zares pomembno, ter kdaj je pripomoček varen in učinkovit. Gre za tako različne medicinske pripomočke, da je uredbo težko zapisati nedvoumno za vse, kaj zahteva in kako jo v praksi lahko izvajamo.«
Zato se pri tem evropska komisija ves čas usklajuje na podlagi vprašanj, ki jih dobi tako od proizvajalcev kot od priglašenih organov, kakor tudi znotraj skupin priglašenih organov, ki imajo svojo koordinacijo. Na podlagi tega nastajajo smernice, ki interpretirajo posamezne zahteve; to pa ponavadi traja najmanj dve leti.
»Evropska komisija (EK) ima svojo strokovno skupino. Predlogi gredo tudi v posvetovanje skupini priglašenih organov. Na koncu o njih EK odloča samostojno in neodvisno. Mehanizmi za interpretacijo so postavljeni, ker je pa uredba tako dolga, je teh interpretacij veliko, saj je veliko tudi medicinskih pripomočkov. Prejšnja direktiva (MDD) je veljala od leta 1993 do 2017 in v teh 24 letih je bilo, mislim, približno pet dopolnil, eno najpomembnejših je bilo leta 2007. Štirinajst let po uveljavitvi direktive so naredili res pomemben korak, ko so na področju dokazovanja klinične učinkovitosti pripomočkov postavili dodatne zahteve tudi za nižje razrede.«
Tudi pri novi uredbi se pričakuje, da bo kar nekaj let potrebnih za razrešitev najtežjih vprašanj. In kot še pravi Ana Pribaković Borštnik, se zaradi vedno novih spoznanj in vse hitrejših tehnologij postavlja vprašanje, kako jih obravnavati – na primer na področju umetne inteligence ali pri uporabi različnih snovi, kjer je meja med zdravilom in medicinskim pripomočkom lahko tanka. In o teh tako imenovanih borderline oziroma mejnih proizvodih se nenehno odloča.
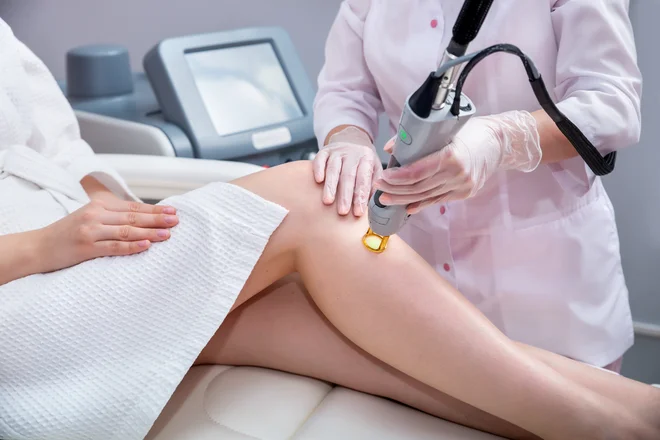
Dandanes se po svetu spopadamo z vse večjo uporabo različnih vsadkov, botoksa in podobnih izdelkov za kozmetične namene. In ali se te tudi preverja glede varnosti? »Niti po direktivi niti po uredbi MDR to niso medicinski pripomočki. Kljub temu je uredba opredelila šest skupin takih pripomočkov, ki sicer nimajo medicinskega namena, vendar jih je zaradi varnosti prav tako treba potrditi. Njihove medicinske učinke se dokazuje skladno s posebnimi akti, ki jih bo za vsako skupino pripomočkov sprejela evropska komisija, torej z nekaj specifičnosti pri dokazovanju delovanja pripomočka s klinično oceno. Dokazovanje varnosti pa mora biti izvedeno na povsem enak način kot pri medicinskih pripomočkih. Gre za botoks, vsadke, različne kožne depilatorje, različna obsevanja, barvne leče in podobno.«
V Nemčiji imajo to že nekaj časa urejeno, saj so dovoljevali proizvajalcu, ki je imel, na primer napravo za odstranjevanje dlak (ki je dejansko kozmetični pripomoček), da je bil ta hkrati pod določenimi pogoji tudi medicinski pripomoček – ker je v določenih primerih odstranjevanje dlak tudi nujni poseg. Pomembno je bilo, da je nad napravami pregled, zelo jasno je moralo biti tudi, katera naprava je in katera ni medicinski pripomoček. »V resnici je enako nevarno v obeh primerih, saj pripomoček za odstranjevanje dlak lahko povzroči opekline pri napačni uporabi. Zanimivo pa je, da v tej skupini ni naprav za izdelovanje tatujev, ampak le naprave za njihovo odstranjevanje.«
Uredba o medicinskih pripomočkih (MDR) velja tudi za skupine izdelkov brez predvidenega medicinskega namena. Uredba zanje velja skoraj enako kot za mediacinske pripomočke, z nekaj specifičnosti pri dokazovanju delovanja pripomočka s klinično oceno.
Teh šest skupin izdelkov so:


Komentarji